
A genetic condition also known as hereditary retinal dystrophy, retinitis pigmentosa (RP) affects the light-sensitive layer of the retina. This is a progressive condition resulting in severe visual loss.
While there is currently no cure for RP, individuals with the condition can maximize the use of their remaining vision with the help of visual aids and rehabilitation (training) programs.
Globally, approximately one out of 5000 people are affected by RP making it the most common inherited disease.
SYMPTOMS:
Night blindness
Having trouble seeing in low light
Progressive decreased side vision leading to tunnel vision
Decreased color perception with degradation of cones
Decreased central vision; in advanced stage
Flashes of light
CAUSE:
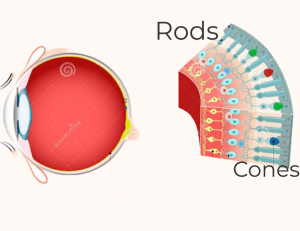
In the retina, light-sensitive photoreceptors are divided into two types: rods and cones. While cones are centrally populated and aid in central vision and seeing in bright light, rods are more densely distributed in the periphery and are responsible for peripheral vision, which includes seeing in the dark.
Rod-cone dystrophy is the most common type of RP, where the retinal rod photoreceptors degenerate and die as a result of biochemical dysfunction brought on by genetic mutation. As a result, night blindness and a gradual loss of peripheral vision ensue.
Some types of RP can primarily affect the cones, causing central vision loss and color disturbances before night vision and peripheral vision loss.
In most cases, RP does not directly affect the cones. Cones are affected when the disease advances and retinal cells continue to degrade, creating a toxic environment.
VISUAL LOSS IN RETINITIS PIGMENTOSA
Most RP patients become legally blind over time. But due to the relatively unaffected cones in the macula, which provide central vision, they rarely become completely blind. In some types of RP though, central vision is affected too, which eventually results in blindness.
The age of onset and inheritance pattern affect the prognosis for patients with retinitis pigmentosa.
RP can be inherited by following patterns:
Autosomal Dominant (AD):
In this type of inheritance pattern, one altered copy of a gene in each cell is sufficient to cause retinitis pigmentosa.
Symptoms usually appear in adulthood only after a person reaches the age of 20.
Autosomal Recessive (AR):
This inheritance pattern for retinitis pigmentosa requires both copies of a mutated gene in each cell.
Each parent of a person with an autosomal recessive condition carries one copy of the mutated gene, but usually, neither parent experiences the disease’s symptoms.
Early onset symptoms, severe vision loss, and night blindness are all symptoms of the autosomal recessive form of RP. Early adolescence is when the symptoms of the autosomal recessive form first appear.
X-linked inheritance:
Genes associated with X-linked retinitis pigmentosa are located on one of the two sex chromosomes, the X chromosome.
The sex chromosome in males have one X chromosome and one altered copy of the gene per cell is enough to cause the condition in them. While females have two X chromosomes and a mutation typically needs to occur in both copies of the gene in them for RP to manifest.
The prognosis and level of vision loss associated with this type of RP are the worst.
Diagnosis
It generally begins with history taking followed by a comprehensive eye examination. This includes tests of visual acuity, visual fields, electroretinography (ERG), and an examination of the retina.
- Colour vision: It is necessary to assess color vision to determine the presence and progression of decreased color perception, which will reveal the extent of cone photoreceptor involvement.
![]()
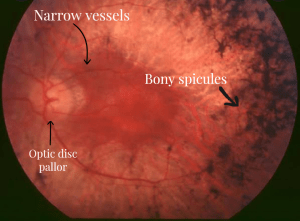
- Retinal examination: Retina is examined using an ophthalmoscope. The presence of bony spicule pigmentation in the retina, narrowing of the retinal vessels, and optic disc pallor are three clinical manifestations that are typical of retinitis pigmentosa. These signs suggest retinal cell death, low metabolic activity in the retina, and poor vision.
- Electroretinography. This examination measures the retina’s electrical activity, or how well the retina reacts to light.
- Visual field testing. The peripheral (side) vision is impacted by RP. A visual field test can help assess the side vision and locate any potential blind spots.
- OCT (Optical Coherence Tomography): This imaging test takes unique, meticulous photographs of the retina. It can be used to identify RP and determine how it affects your retina.
- Genetic analysis: This test examines a sample of your blood or other tissues to determine whether you possess specific genes related to a particular disease type. The severity of the disease may also be predicted using this data, as well as whether gene therapy to replace the problematic gene might be beneficial.
IS RETINITIS PIGMENTOSA CUREABLE?
Retinitis pigmentosa currently has only conservative treatments available to help slow the disease’s progression. However, there are currently clinical trials taking place for some new treatments that appear promising.
Since over 100 different genes can cause RP, there is no single cure. Researchers are looking into the causes and mechanisms of RP in families. Based on this knowledge, they intend to develop treatments.
Rod-cone dystrophy, retinitis punctata albescens, and Usher syndrome are a few of the various types of RP.
There is now medication available to treat one type of RP caused by an RPE65 gene defect. The drug is injected into the retina during a vitrectomy procedure.
Different genetic defects linked to different types of RP make it difficult for researchers to develop a single treatment for them all.
Vitamin A supplement:
RP patients previously received vitamin A and antioxidant supplements to improve their retinal health.
There is a growing concern that a high intake of Vitamin A supplements can cause overdose leading to liver toxicity. Furthermore, there isn’t much evidence that it slows down the progression of RP.
Your ophthalmologist can give you advice on the advantages and disadvantages of vitamin A as well as how much you can safely consume.
Low vision aids:

Currently, there are tools and apps to help patients with low vision to enhance vision. Examples of these tools include telescopic glasses, magnifiers, AR glasses, etc. With the help of vision experts, people with low vision can learn how to use these aids and make the most of their remaining vision.
Cataract removal surgery:
In retinitis pigmentosa, cataract is a significant secondary cause of vision impairment. An early onset posterior subcapsular cataract is the most common morphological type described in the literature.
According to a study, most patients with RP experience a significant improvement in visual acuity following surgical removal of cataract. After surgery, the burden of vision loss and blindness significantly decreases. Patients who arrive on time and have useful preoperative vision are probably to have better postoperative visual results.
FUTURE TREATMENT OF RP:
Some future treatments for RP include gene therapy and stem cell transplants and artificial retinal implants.
Gene therapy
It is a treatment that replaces the defective gene that causes RP with a healthy gene.
The retina receives healthy copies of the defective gene via an injection during vitreoretinal surgery.
For the treatment of different types of RP, researchers are experimenting with gene therapy. This includes clinical trials for X-linked retinitis pigmentosa, retinitis pigmentosa caused by a mutation in the RHO gene, retinitis pigmentosa with Usher syndrome caused by the mutation in the USH2A gene, etc.
Consult your ophthalmologist to learn more about clinical trials and to find out if you qualify to take part.
Stem cell transplant
This is a treatment that replaces the damaged cells in the eye with healthy cells. The safety and efficacy of these treatments are being tested in clinical trials.
Artificial retinal implants:

A tiny electronic device is surgically implanted in the retina. Patients must also put on a specific pair of glasses with video processing technology. Through the glasses, light is transmitted to the device, which then activates the light-sensitive cells in the retina and sends the images to the brain.
There is hope that one or more of these treatments will be effective in treating RP and slowing the progression of vision loss
GenSight may benefit all types of retinitis pigmentosa
GenSight Therapeutics uses an optogenetic method involving an eye injection and cutting-edge goggles in the treatment of all types of RP.
A gene is injected into retinal cells to help them react to light. These cells communicate with the brain by sending electrical signals through the goggles.
The injection and the goggles try to mimic the function of photoreceptors, which are light-sensing cells that don’t function properly in people with retinitis pigmentosa.
GENETIC COUNSELLING:

Genetic counseling can assist patients with retinitis pigmentosa, a hereditary condition, in comprehending the risks and deciding whether to have children.
If you have RP and want to know if you will pass this eye condition on to your children, speak with a genetic counselor.
PSYCHOLOGICAL IMPACT

One of the most common changes in patients with RP and associated vision loss is depression. People with RP frequently distance themselves from friends and family and are more likely to experience depression.
Friends and family must be understanding and supportive as the disease worsens. They should also assist the person in becoming independent despite their vision loss by assisting them in adjusting to low vision aids.
It is necessary to closely monitor and supportively manage such patients to identify depression and treat it as soon as possible.